Government’s Drug Discovery Hackathon: A Game Changer in Open Innovation Model

There are many innovation challenges launched by the Indian government in the past few years and one of the high-level competitions is to discover a drug. Drug discovery is a big term for a larger population but the Government of India’s Drug Discovery Hackathon has the power to simplify this high demanding requirement due to COVID-19 situations that can result in many professionals participating in the Hackathon.
Coordination and duplicacy are some of the main factors for the contest to be successful since multiple participants will have similar ideas and reconciliation is a must to avoid duplicate efforts bring every entry into the system is itself a challenge. Great coordination already planned as the Hackathon is scheduled in the phased manner. Portals created for such competitions are capable of handling different types of data.
There are three phases of the Hackathon spanning 2 months each with Phase 1 will close accepting entries on 30th September 2020.
Phase 1: Screening
Phase 2: Improvisation
Phase 3: Finalization
Indian students across the world, who have an Indian passport, have a big opportunity to display their potential.
A list of Problem Statements given on the mygov.in website noted below:
There are three tracks with multiple problem list
Track 1: COVID Specific Drug Discovery, a total of 15 problems
Track 2: General Drug Discovery, Including COVID, a total of 14 problems
Track 3: Moon Shot Approach
Below the video is the toolkit for the problem list:
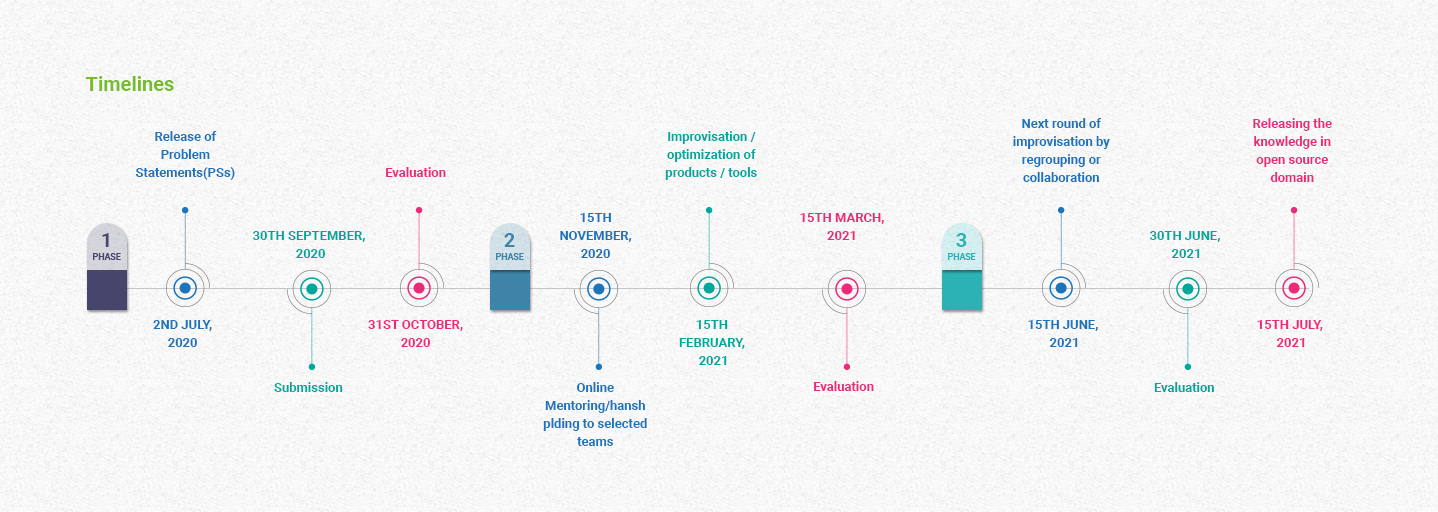
Timelines From MyGov.in Website
FAQs for Hackathon
-
- Computer Sciences
- Pharmaceutical Sciences
- Biotechnology
- Machine Learning
- Artificial Intelligence
- Drug Discovery
- In Silico Chemistry
- Big Data Analytics
-
- 3th September 2020
-
- A joint initiative of AICTE, CSIR, and supported by Office of Principal Scientific Advisor, Govt. of India, NIC and MyGov.
-
- In-Silico Drug Discovery
- Chemical synthesis
- and Biological Testing
-
- Chemical Compounds,
- Bioactivity,
- Bioavailability,
- Host-pathogen Interaction Maps, and
- Several Algorithms for prioritization of known approved compounds or discovery of new ones
-
- Developing a regression-based model for screening compounds with 3CLpro inhibitory activity
- Fragment-based de novo design of exemplar inhibitor against SARS-CoV-2 spike glycoprotein
- Screening of medicinal plants compound datasets against multiple targets of SARS-COV-2
- Computational screening of molecular fragments and fragment linking to design novel inhibitors for SARS-Cov-2 main protease
- Designing newer quinoline analogues by performing molecular docking analysis of hydroxychloroquine and other quinolone agents against Spike- ACE2 complex, Transmembrane protease serine 2 and Spike protein of SARS-CoV-2
- Development of nucleotide analogs library by performing virtual screening using molecular docking methodologies at the active site of RNA dependent RNA polymerase enzyme of SAS-COV-2
- Identifying covalent spike protein inhibitors to control the infection of SARS-CoV-2 from small molecule databases
- Computational screening to identify lead drug candidates binding to NSP12 [RNA dependent RNA polymerase (RdRp)] from SARS CoV-2
- Computational screening of synthetic/natural compound databases to identify Furin enzyme inhibitors and in silico prediction of their toxicity potential in host
- Targeting the N- and C-terminal of nucleocapsid protein of SARS-CoV-2 for identification of small molecules through virtual screening
- To design small molecules binding to glycan groups on the Spike protein on the surface of SARS-Cov2
- Construction of homology models of wild and mutant D614G spike protein
- Identification and characterization of potent inhibitors by exploring the interaction interfaces between Envelope protein of SARS-CoV-2 and host protein –H2A and BRD2/4 (Dr.Shailendra Asthana)
- Probing helminth proteome for anti-hypertensive small natural peptides (small peptide of <2000 kDa) inhibitor for ACE2/ Cathepsin L to inhibit SARS-CoV-2 entry
- Building a complete structural model for SARS-CoV-2
-
- Develop a reinforcement learning-based algorithm to identify lead molecules by emulating ligand-protein interactions
- Machine learning models to prioritize optimal parameters of predicted ADME and Toxicity data
- Building SARS-CoV-2 Inhibitor Knowledgebase – SAVIOR
- Machine intelligence design and development of main protease inhibitors drugs.
- To develop a regression based QSPR model for Caco-2 cell permeability.
- Predicting the biological properties of potential SARS-CoV-2 inhibitors using graph theory and machine learning
- ML model to predict small molecule clinical trial success probability by phase
- Matched Pair Analysis based tool to generate isosteric and bioisosteric molecular libraries
- Automated Analogue library generation tool using bioisosteres
- ML model for antiviral peptide predictions using Generative Adversarial Networks
- Implement a framework to mine protein-ligand & protein-protein interaction networks for drug repurposing
- To improve the efficiency of MOLS algorithm in terms of sampling, scoring and computational time
- Developing a linear discriminant analysis model for screening pharmaceutical compounds with hERG inhibitory activity (cardiotoxicity) and using the model to screen CAS antiviral database to identify compounds with cardiotoxicity potential
- A computational pipeline to predict Drug Induced Liver Injury (DILI).
For more details and toolkit videos about Problem Statements see here
For complete details of the Durg Discovery Hackathon see here